Author: Denis Avetisyan
Researchers have developed a method to significantly reduce the computational demands of accurate electronic structure calculations without sacrificing precision.
This work introduces an active space Unitary Coupled Cluster method that balances internal and external correlation using UCCSD(4)/MP2, demonstrating improved efficiency across diverse chemical systems.
Despite its promise for accurately describing strongly correlated systems, the computational cost of Unitary Coupled Cluster (UCC) theory often limits its practical application. This work, ‘Reducing the Cost of Unitary Coupled Cluster via Active Space Partitioning’, introduces an efficient UCCSD(4)/MP2 approach that leverages an active space treatment of internal correlation coupled with a perturbative description of external excitations. The resulting methodology, tested across diverse chemical systems, demonstrates that accurate potential energy surfaces can be obtained with significantly reduced computational effort-using as few as 15-25% of the virtual orbitals-while maintaining robustness in orbital basis choice. Will this active space framework enable accurate and scalable electronic structure calculations for increasingly complex chemical systems on both classical and quantum computers?
The Persistent Challenge of Electron Correlation
The accurate depiction of electron correlation-the complex interplay between electrons in a multi-electron system-forms a cornerstone of quantum chemistry, yet presents a persistent computational hurdle. While the Schrödinger equation precisely describes these interactions, its solutions are intractable for all but the simplest atoms and molecules. Traditional methods, like Hartree-Fock, often treat electron correlation as a perturbation, introducing approximations that can become severely limiting when electrons are strongly interacting. These approximations frequently fail to capture the true behavior of electrons, particularly in scenarios involving bond breaking or the description of excited states, leading to qualitatively incorrect predictions of molecular properties and reaction outcomes. The computational cost of methods that do attempt to accurately account for electron correlation-such as configuration interaction or coupled cluster-scales dramatically with system size, restricting their application to relatively small molecules and hindering the study of larger, more complex chemical systems.
The behavior of electrons in multi-electron systems isn’t simply the sum of their individual actions; rather, intricate many-body effects arise from their mutual interactions. Accurately modeling these interactions is crucial, but often requires approximations due to the computational expense of a full solution. When ‘static correlation’ – the strong, simultaneous interaction between multiple electrons – dominates, these approximations can falter dramatically. This failure isn’t merely a matter of reduced precision; it can lead to qualitatively incorrect predictions about molecular structure, bonding, and reactivity. For instance, a system might be predicted to have a single stable configuration when, in reality, it exhibits multiple stable states or even dissociation. These failures highlight the need for computational methods that can reliably capture strong static correlation, particularly in systems undergoing bond breaking or forming.
Computational chemistry continually strives to accurately model the interactions between electrons within molecules, a task complicated by the phenomenon of electron correlation. While methods exist to account for dynamic correlation – the instantaneous interactions arising from electron movement – simultaneously and efficiently capturing static correlation, which occurs when multiple electronic configurations contribute significantly to the overall wavefunction, remains a formidable hurdle. This difficulty stems from the exponential scaling of computational cost with system size when attempting to explicitly include all relevant configurations; approximations are therefore essential, but often struggle to balance accuracy and feasibility. Consequently, developing methods capable of efficiently addressing both forms of correlation is central to accurately simulating chemical processes, predicting molecular properties, and ultimately, designing new materials and reactions.
Computational chemistry routinely encounters a fundamental tension between the desire for highly accurate predictions and the practical limitations of available computing resources. Many chemical systems, especially those involving transition metals, strong correlation, or multiple interacting electronic states, present a significant challenge to standard quantum chemical methods. While techniques like Density Functional Theory (DFT) offer a reasonable compromise for many applications, they can falter when dealing with complex electronic structures where electron correlation is crucial. More sophisticated methods, such as Coupled Cluster theory, provide high accuracy but scale factorially with system size, rendering them intractable for all but the smallest molecules. Consequently, researchers constantly seek methods that offer a better balance – algorithms capable of capturing essential correlation effects without incurring prohibitive computational costs, particularly when simulating chemical reactions or the properties of large molecules.
Strategic Simplification: The Active Space Approach
Electronic structure calculations scale poorly with system size, primarily due to the exponential growth in the number of orbitals requiring treatment. To mitigate this, the ‘Active Space’ approach selectively correlates a subset of molecular orbitals, typically those most important for describing the electronic behavior of the system. This reduces computational cost by excluding high-lying or core orbitals that contribute little to the overall energy and properties. The selection of orbitals for inclusion in the active space is critical; commonly, these are the highest occupied and lowest unoccupied molecular orbitals, but more sophisticated methods exist to optimize this choice based on orbital energies, occupation numbers, or contributions to the total energy. By focusing computational effort on the active space, larger systems become tractable while retaining a reasonable level of accuracy.
Unitary Coupled Cluster Singles and Doubles with fourth-order truncation, denoted UCCSD(4), serves as a high-accuracy benchmark within electronic structure calculations. This method builds upon the coupled cluster (CC) theory by systematically incorporating single and double excitations from a reference determinant, and includes perturbative corrections accounting for fourth-order triple and higher excitations. UCCSD(4) provides a significantly improved treatment of electron correlation compared to lower-level CC methods like CCSD, and approaches the accuracy of the full CC hierarchy, CCSDT, while remaining computationally more tractable. Its robustness stems from its size-extensive properties and well-defined convergence behavior, enabling reliable assessments of approximations used in more efficient, albeit less accurate, computational schemes. The resulting energies and properties from UCCSD(4) calculations are frequently used as ‘gold standard’ values against which the performance of density functional theory (DFT) and other reduced-cost methods are evaluated.
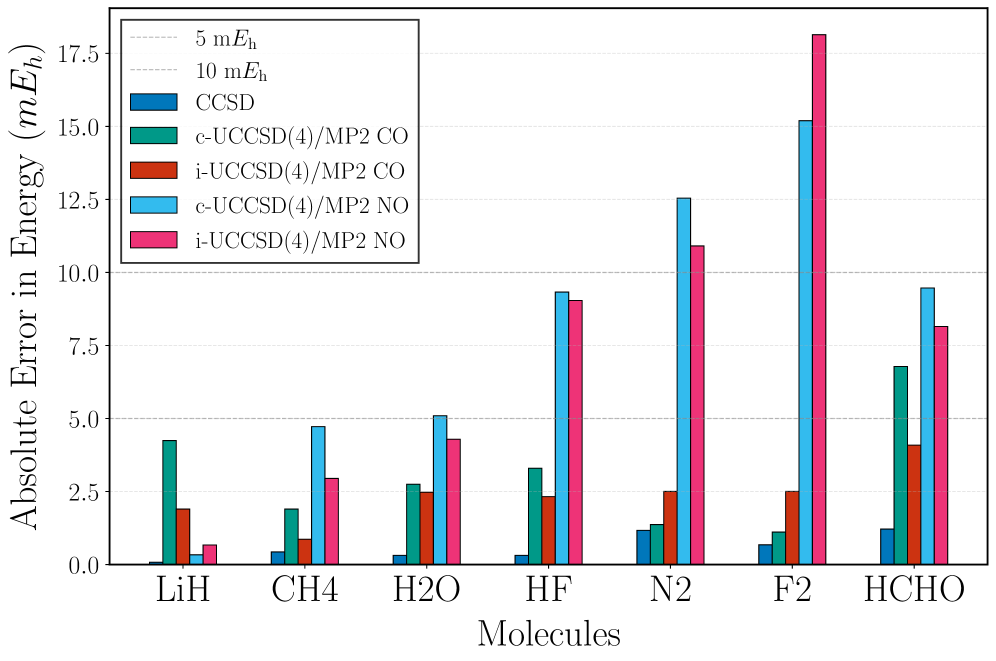
Unitary Coupled Cluster Singles and Doubles with fourth-order truncation (UCCSD(4)) effectively models dynamic correlation, accounting for electron interactions that significantly impact molecular properties. However, the computational cost of UCCSD(4) scales factorially with system size – approximately N^8 where N represents the number of orbitals – limiting its direct application to systems exceeding a few hundred electrons. Furthermore, UCCSD(4) struggles with strong static correlation, arising from near-degeneracy in the electronic structure, requiring excessively large basis sets or specialized techniques to achieve accurate results in such cases. This computational demand necessitates the use of approximations or reduced active spaces for larger systems, potentially compromising accuracy.
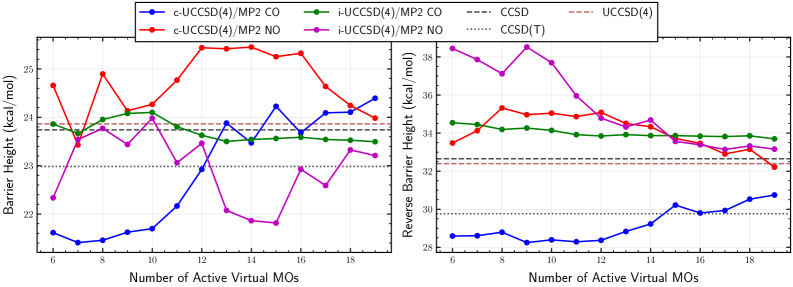
Selecting an appropriate set of orbitals for the active space in electronic structure calculations is fundamental to achieving a balance between computational cost and accuracy. The number of orbitals included directly impacts the scaling of the calculation; therefore, minimizing this number without sacrificing essential physics is critical. ‘Frozen Natural Orbitals’ represent a technique for compacting this representation by identifying and excluding orbitals that contribute minimally to the overall correlation energy. These orbitals are determined via a natural orbital transformation of the one- and two-electron density matrices, effectively providing a basis set optimized for describing the dominant correlation effects and reducing the size of the active space without significantly impacting the final result. This approach leverages the fact that not all orbitals contribute equally to the electronic correlation, allowing for a more efficient and targeted calculation.
Synergistic Precision: Coupling UCCSD(4) and MP2
Hybrid quantum chemical methods combine the strengths of different computational approaches by partitioning the molecular system into active and external spaces. Specifically, UCCSD(4) – a variant of coupled cluster theory – is applied to the actively correlated electrons within a defined active space, while Møller-Plesset perturbation theory (MP2) is employed to treat the remaining, less critical, electrons in the external region. This partitioning allows for a reduction in computational scaling; UCCSD(4) typically scales as N^6, where N is the number of basis functions, but by limiting its application to the active space and supplementing with MP2 for the external space, the overall computational cost can be significantly decreased.
Hybrid approaches combining UCCSD(4) and MP2 are implemented through two distinct strategies. The ‘Composite’ method performs a UCCSD(4) calculation within the active space, followed by a separate MP2 calculation on the external space, with no interaction between the two results. Conversely, the ‘Interacting UCCSD(4)/MP2’ approach allows for full coupling between the UCCSD(4) and MP2 calculations, meaning the MP2 treatment directly incorporates the UCCSD(4) active space results during perturbation theory, enabling a more holistic and potentially accurate, but computationally intensive, treatment of electron correlation.
The combination of UCCSD(4) and MP2 in a coupled approach yields significant computational savings by exploiting the differing strengths of each method. UCCSD(4) accurately describes correlation within the active space, while MP2 efficiently handles the external, less critical regions of the wavefunction. This partitioning allows for a reduction in computational cost – up to two orders of magnitude compared to performing a full UCCSD(4) calculation on the entire system – because MP2’s scaling with system size is lower than that of UCCSD(4). The overall efficiency is achieved by treating only the active space with the computationally demanding UCCSD(4) method, and applying MP2 to the remaining, larger, inactive space.
Hybrid UCCSD(4)/MP2 methods achieve a balance between computational cost and accuracy by strategically partitioning the electronic correlation problem into active and external spaces. The active space, encompassing the most critical correlation effects, is treated with the more computationally demanding UCCSD(4) method, while the external space is addressed with the less expensive MP2. This partitioning, dependent on careful selection of the active space, significantly reduces the overall computational expense compared to performing UCCSD(4) on the entire system, while maintaining a high degree of accuracy for diverse chemical systems, including those with strong correlation effects.
Validation and Impact: A New Standard of Accuracy
The GW100 dataset represents a significant advancement in the validation of computational chemistry methods, offering a standardized and challenging benchmark for assessing their predictive power. This carefully curated collection comprises 100 diverse molecular structures, each with highly accurate reference energies and geometries calculated using sophisticated quantum chemical techniques. By subjecting various electronic structure methods to rigorous testing against the GW100 benchmark, researchers can objectively evaluate their ability to accurately predict molecular properties, identify limitations, and guide further methodological development. The dataset’s breadth, encompassing a range of chemical environments and molecular sizes, ensures that evaluations are not specific to a narrow class of compounds, offering a comprehensive measure of a method’s overall performance and reliability in predicting both ground-state energies and molecular shapes.
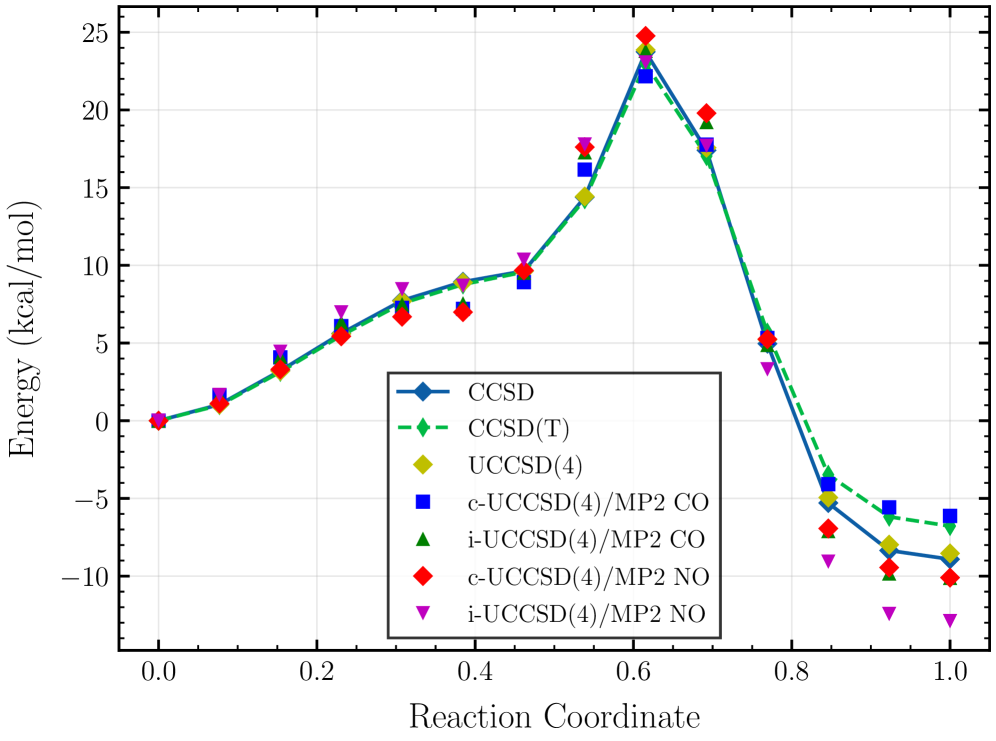
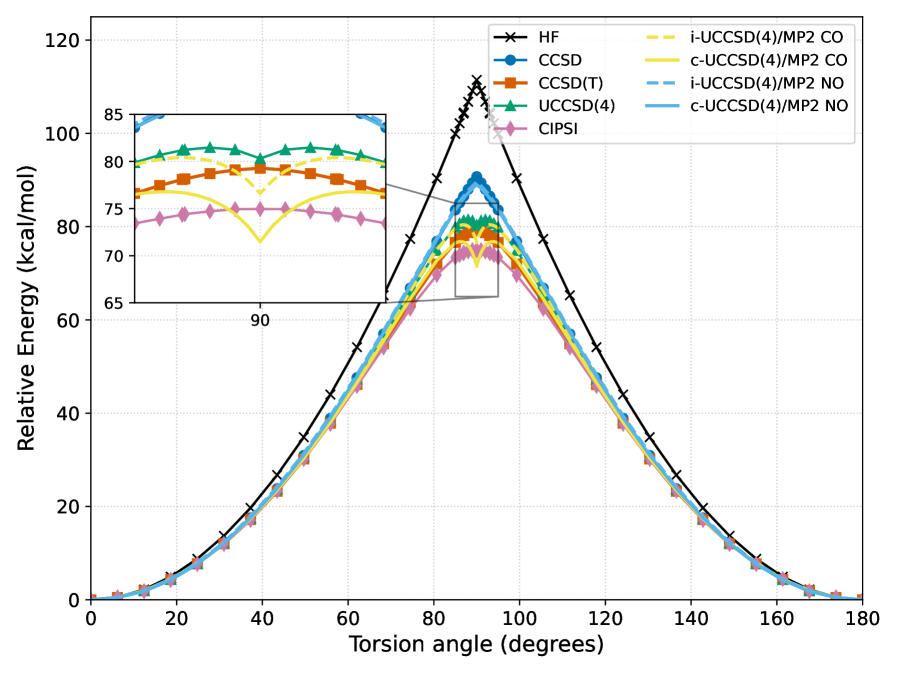
The ethylene torsion benchmark serves as a crucial test for evaluating how well computational methods account for static correlation – a phenomenon arising when multiple electronic configurations contribute significantly to the overall wavefunction. Traditional methods often struggle with this, leading to inaccurate predictions for systems where a single electronic configuration is insufficient. Recent studies employing advanced techniques on this benchmark demonstrate a marked improvement in capturing these subtle electronic interactions. By accurately modeling the energy changes as ethylene rotates, these methods prove their capability to go beyond single-reference approximations and provide a more reliable description of chemical bonding and reactivity, particularly in scenarios involving bond breaking and formation where static correlation is prominent.
Investigating phosphate hydrolysis, frequently used as a simplified model for the biologically crucial process of ATP hydrolysis, rigorously tested the methods’ capacity to accurately describe chemical reactions involving bond scission and formation. This demanding application requires precise treatment of changes in electronic structure as bonds break and new ones emerge – a significant challenge for many electronic structure methods. The successful modeling of phosphate hydrolysis demonstrates the hybrid UCCSD(4)/MP2 approach’s ability to not only determine stable molecular geometries and energies, but also to accurately track the energetic landscape of a chemical reaction, providing insights into reaction mechanisms and rates. This capability extends the method’s utility beyond static structural predictions and into the dynamic realm of chemical processes, highlighting its potential for studying complex biochemical reactions.
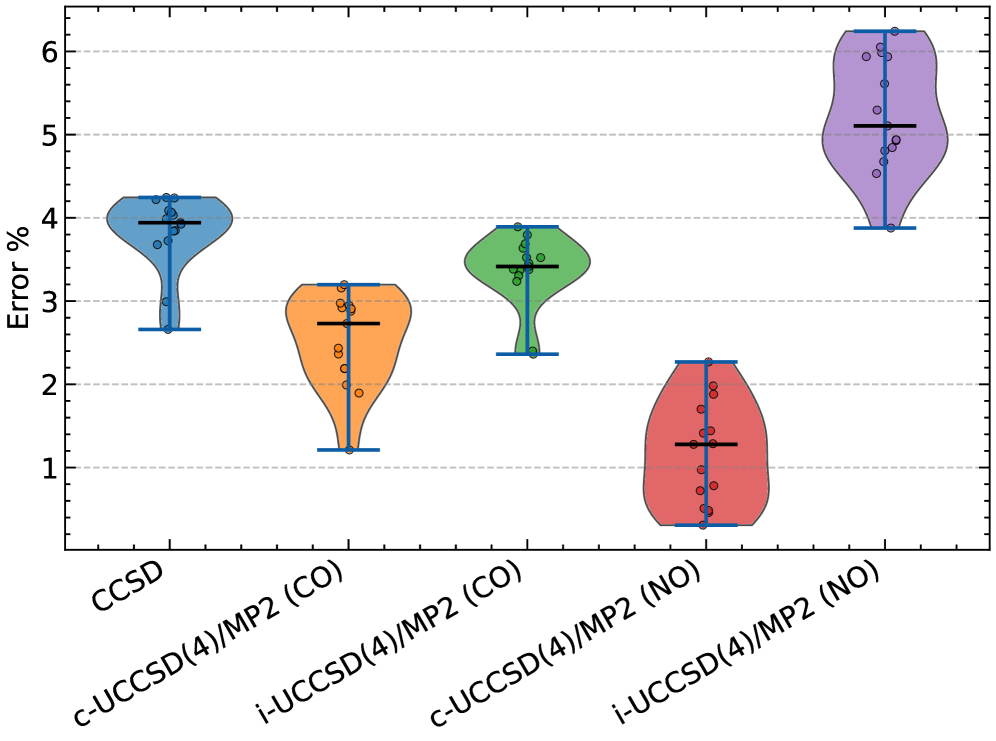
Rigorous testing of electronic structure methods using the GW100 dataset revealed a particularly strong performance from the Composite UCCSD(4)/MP2 method, enhanced with natural orbitals. This approach achieved a mean absolute error of just 1.5% in predicting the energies and geometries of molecules within the medium-sized GW100 set – demonstrably the lowest error rate among all methods evaluated. This level of accuracy signifies a substantial advancement in computational chemistry, offering a reliable pathway to model complex chemical systems with unprecedented precision and efficiency. The method’s success stems from its ability to effectively balance computational cost with high-level correlation effects, making it a valuable tool for a wide range of applications, from materials science to drug discovery.
Rigorous validation studies demonstrate the hybrid UCCSD(4)/MP2 approach as a dependable and computationally efficient pathway toward high-accuracy electronic structure calculations. Performance benchmarks, including assessments on the GW100 dataset and the challenging ethylene torsion benchmark, consistently reveal its ability to accurately predict molecular energies and geometries, even in scenarios demanding precise treatment of static correlation. Further application to phosphate hydrolysis-a simplified model for the vital biological process of ATP hydrolysis-confirms its competence in describing chemical reactions involving bond breaking and formation. Notably, on the GW100 medium-sized molecule set, this composite method, leveraging natural orbitals, achieved a mean absolute error of only 1.5%, establishing it as a leading performer among currently available techniques and solidifying its potential for broad application in diverse chemical and materials science investigations.
The pursuit of computational efficiency in quantum chemistry, as demonstrated by this active space partitioning of Unitary Coupled Cluster, echoes a fundamental principle of scientific rigor. It isn’t about achieving a single, elegant solution, but relentlessly refining approximations. As Ernest Rutherford observed, “If you can’t explain it simply, you don’t understand it well enough.” This work exemplifies that sentiment; it doesn’t shy away from the inherent complexity of electronic structure, but seeks to manage it through strategic partitioning – treating internal and external correlation with distinct, yet complementary, methods. The choice of orbital space, crucial to the method’s success, isn’t about finding the ‘right’ answer immediately, but about iterative improvement through controlled approximations. The more visualizations, the less hypothesis testing, indeed.
Where Do We Go From Here?
The partitioning of electronic correlation, as demonstrated by this work, offers a predictable benefit: a compromise between the rigorously complete, and the computationally tractable. The UCCSD(4)/MP2 scheme, while promising, merely shifts the burden of accuracy from one place to another. The ‘optimal’ balance between internal and external correlation will, predictably, vary with the system under investigation – and, more subtly, with the definition of ‘cost’ itself. Is it wall-clock time? Energy consumption? The patience of the researcher?
The sensitivity to orbital choice, while acknowledged, hints at a deeper issue. Frozen Natural Orbitals, and active space selection generally, represent a pragmatic limitation, but they introduce an implicit bias. Future work must address the systematic quantification of this bias – and, more challengingly, explore methods for mitigating it without negating the computational gains. One suspects that a truly robust approach will require moving beyond the single-reference framework altogether, despite the attendant complexities.
Ultimately, this work is a step along a familiar path. Models are, after all, not reflections of truth, but convenient fictions. The value of this particular fiction lies not in its absolute accuracy, but in its ability to consistently outperform its predecessors – until, inevitably, it too is surpassed. The pursuit of electronic structure theory is, therefore, less a quest for perfection and more a disciplined exercise in controlled approximation.
Original article: https://arxiv.org/pdf/2602.04783.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- All Skyblazer Armor Locations in Crimson Desert
- How to Get the Sunset Reed Armor Set and Hollow Visage Sword in Crimson Desert
- All Shadow Armor Locations in Crimson Desert
- Marni Laser Helm Location & Upgrade in Crimson Desert
- All Helfryn Armor Locations in Crimson Desert
- All Golden Greed Armor Locations in Crimson Desert
- All Icewing Armor Locations in Crimson Desert
- Best Bows in Crimson Desert
- How to Beat Stonewalker Antiquum at the Gate of Truth in Crimson Desert
- Legendary White Lion Necklace Location in Crimson Desert
2026-02-05 19:16