Author: Denis Avetisyan
A novel approach leveraging density functional theory as a reference point dramatically improves the reliability of coupled cluster calculations for challenging systems, particularly those containing transition metals.
Kohn-Sham density encoding provides a superior starting point for coupled cluster theory, enhancing accuracy for molecules exhibiting strong multireference character.
Despite the established accuracy of coupled cluster theory, its performance falters for strongly correlated systems, often necessitating computationally expensive multireference approaches. This research, presented in ‘Kohn-Sham density encoding rescues coupled cluster theory for strongly correlated molecules’, reveals that employing a Kohn-Sham density functional theory (KS-DFT) reference dramatically improves coupled cluster calculations by encoding crucial information within the one-particle density matrix. Specifically, KS-CCSD(T) achieves near-chemical accuracy for challenging systems-including the notoriously difficult Cr_2 potential energy surface-and provides a diagnostic to assess multireference character. Could this approach unlock practical, accurate treatment of strong correlation within the widely-used coupled cluster framework and accelerate materials discovery and machine learning potential development?
Unraveling the Complexity: Transition Metal Electronic Structure
Transition metal compounds present a unique modeling challenge in computational chemistry stemming from their inherent multireference character – a situation where the electronic structure cannot be adequately described by a single electronic configuration. Unlike many simpler molecules, these compounds exhibit strong electron correlation, meaning the behavior of one electron is heavily influenced by all others, necessitating the consideration of multiple electronic configurations simultaneously. This multireference nature arises from the partially filled d and f orbitals characteristic of transition metals, leading to significant static correlation that traditional single-reference methods, such as Hartree-Fock and many coupled cluster approximations, struggle to capture accurately. Consequently, predicting properties like magnetic moments, reactivity, and spectroscopic behavior requires computationally expensive methods capable of accounting for these complex electronic correlations, limiting the size and complexity of systems that can be reliably modeled and hindering advancements in fields reliant on understanding these materials.
Conventional computational approaches, such as Hartree-Fock Theory and Coupled Cluster Theory, frequently encounter limitations when applied to transition metal compounds due to the intricate nature of electron correlation within these systems. These methods, while successful for many chemical scenarios, often treat electron interactions in a simplified manner, failing to adequately describe the simultaneous influence of multiple electrons on each other. Transition metals exhibit a large number of nearly degenerate electronic states, necessitating a more nuanced treatment that accounts for the collective behavior of electrons – a challenge for methods inherently designed for single-reference systems. Consequently, predictions of properties like magnetic moments, reactivity, and spectral characteristics can be significantly inaccurate, hindering advancements in fields reliant on precise understanding of these complex materials.
The inability to accurately model transition metal compounds presents a considerable obstacle to advancements in several applied fields. Precise prediction of catalytic activity, crucial for designing more efficient industrial processes and sustainable chemical transformations, is hampered by the limitations of current computational methods. Similarly, the development of novel materials with tailored properties – from superconductors to advanced alloys – relies on understanding the complex interplay of electronic structure and material behavior, a connection presently obscured by modeling inaccuracies. Perhaps most critically, the function of metalloenzymes – biological catalysts essential for life – depends on subtle electronic effects within the metal center; therefore, a refined understanding of these systems is vital for unraveling biological processes and developing new therapeutic interventions, all of which are currently restricted by the challenges in accurately representing their electronic structure.
Predicting the behavior of transition metal compounds hinges on a comprehensive understanding of their electronic structure, a task complicated by inherent multireference character and strong electron correlation. These compounds don’t conform to the simple electronic descriptions that work well for many molecules; instead, multiple electronic configurations contribute significantly to the overall wave function. Consequently, accurately modeling their properties-reactivity, spectra, magnetic moments-requires computational methods capable of capturing these complex interactions. Researchers are actively developing and refining techniques, such as density functional theory with advanced functionals and multireference configuration interaction, to move beyond the limitations of traditional approaches and unlock the potential for de novo design of materials, catalysts, and metalloenzymes with tailored properties. The ability to reliably predict behavior from electronic structure promises advancements across chemistry, physics, and biology.
Bridging Accuracy and Efficiency: The KS-CC Method
The KS-CC method represents a hybrid electronic structure approach designed to balance computational efficiency with high accuracy. It achieves this by utilizing Kohn-Sham Density Functional Theory (KS-DFT) to generate a reference wavefunction and one- and two-electron integrals, which are then employed within a Coupled Cluster (CC) calculation. This strategy significantly reduces the computational cost typically associated with traditional CC methods, which scale steeply with system size. By leveraging the relatively low cost of KS-DFT to establish a reasonable starting point, KS-CC aims to provide CC-level accuracy for systems where full CC calculations are impractical, particularly for transition metal complexes and other challenging chemical scenarios.
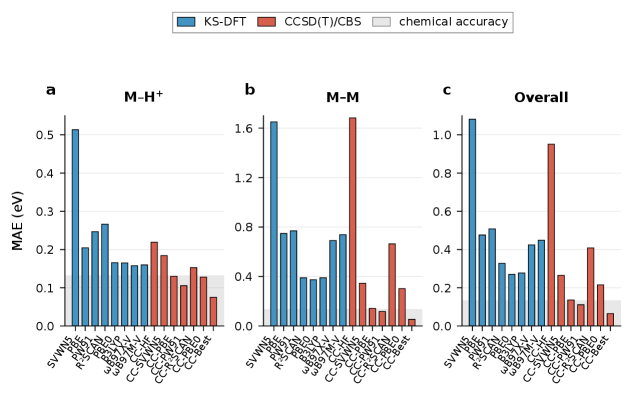
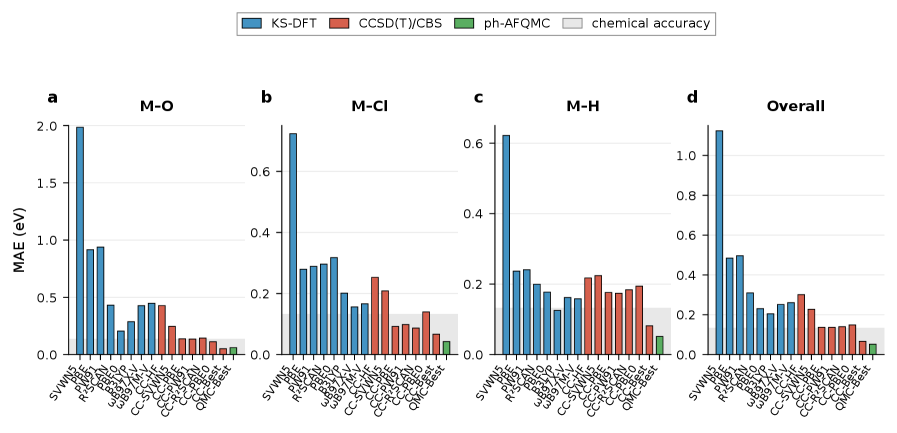
The KS-CC method enhances the accuracy of transition metal thermochemical calculations by utilizing Kohn-Sham Density Functional Theory (DFT) as a reference for subsequent Coupled Cluster (CC) computations. Evaluations demonstrate a mean absolute error (MAE) of 0.05 eV when calculating metal-O, Cl, and H bond dissociation energies. This level of accuracy represents a significant improvement over traditional DFT methods for these systems, particularly those involving transition metals, where electron correlation effects are substantial and often poorly described by standard functional approximations. The method’s success is attributed to the ability of the KS-DFT reference to provide a reasonable starting point for the more accurate, but computationally expensive, CC treatment.
Traditional Coupled Cluster (CC) calculations, while highly accurate, are computationally expensive, particularly when starting from a poor initial wavefunction. The KS-CC method mitigates this cost by utilizing the wavefunction obtained from Kohn-Sham Density Functional Theory (KS-DFT) as the reference for subsequent CC calculations. KS-DFT provides a reasonable single-reference description, meaning the initial wavefunction is already relatively close to the true ground state, thereby significantly reducing the number of iterations and computational resources required for convergence in the CC calculation. This is especially advantageous for systems where traditional ab initio methods struggle to converge or require excessively large basis sets, offering a pathway to high-accuracy calculations at a lower computational cost.
The KS-CC method utilizes a non-canonical Fock operator, differing from traditional Hartree-Fock and Coupled Cluster implementations which employ a canonical form. This non-canonical formulation arises from the use of Kohn-Sham orbitals in constructing the Coupled Cluster excitation operator. Consequently, direct application of standard Coupled Cluster procedures is inaccurate. Achieving reliable results with KS-CC necessitates specialized techniques, most prominently Semi-Canonicalization. This procedure transforms the non-canonical Fock operator into a semi-canonical form, enabling the correct evaluation of the CC excitation energies and ensuring accurate calculation of properties like bond dissociation energies and thermochemical data. Failure to implement Semi-Canonicalization leads to significant errors in the calculated energy levels and molecular properties.
Dissecting the Difference: Validating KS-CC with Density Analysis
Analysis of difference density, calculated as the disparity between electron densities obtained from Kohn-Sham Density Functional Theory (KS-DFT) and KS-CC methods, offers a direct assessment of the impact of incorporating coupled-cluster effects on the electronic structure. This approach allows for the identification of regions in space where the approximations inherent in standard KS-DFT significantly deviate from the more accurate coupled-cluster treatment. By visualizing and quantifying these differences, researchers can pinpoint specific deficiencies in the functional or basis set used, leading to improved methodologies and a more reliable description of chemical systems. The magnitude and spatial distribution of the difference density provide insights into the nature of the electronic correlation effects captured by KS-CC, and can be used to validate the accuracy of the method for specific chemical properties.
Natural Orbital Decomposition (NOD) analyzes the difference density – the variation between electron densities calculated using different computational methods – by expressing it as a sum of contributions from individual natural orbitals. This decomposition identifies the specific orbitals that contribute most significantly to the density difference, thereby pinpointing the regions of electronic structure where the KS-CC method demonstrates improvement or deficiency compared to the reference calculation. By isolating these key orbitals, researchers can determine if KS-CC accurately captures static correlation effects, or if further refinements are needed to better describe the electronic behavior of the system under investigation. The magnitude and spatial distribution of the contributions from each natural orbital provide quantitative insight into the performance of KS-CC in specific chemical environments.
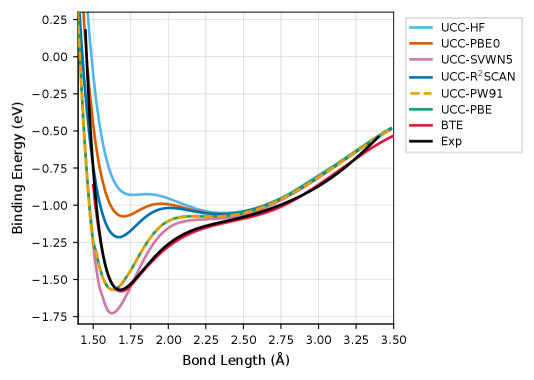
The Density Difference Metric (DDM) quantitatively assesses the discrepancy between electron densities calculated using different computational methods. For the Cr2 dimer, a Normalized Non-local Density Difference (NNED) of 0.083 was measured. This value signifies a substantial alteration in the calculated electron density when employing the KS-CC method compared to standard Density Functional Theory (DFT). A non-negligible NNED indicates that the KS-CC approach introduces a meaningful change in the electronic structure, justifying the observed improvements in computational accuracy for systems involving transition metals; values exceeding a certain threshold are considered evidence of a robust methodological advancement.
Application of the KS-CC method to the Cr2 dimer, utilized as a benchmark system, demonstrates its capability in accurately modeling transition metal compounds. Analysis of the Potential Energy Surface revealed a reduction in error for the dissociation energy (De) to 0.001 eV, and for the equilibrium bond length (re) to 0.04 Å. These results indicate a significant improvement in computational accuracy when modeling systems containing transition metals, validating the method’s potential for broader application in this field.
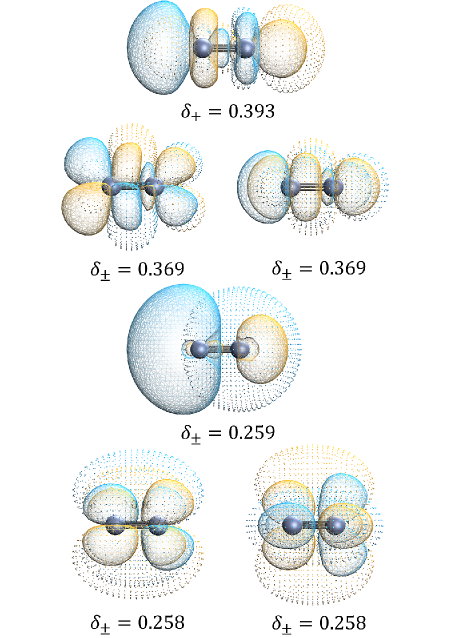
Beyond Current Limits: Implications and Future Trajectories
The enhanced precision in modeling transition metal compounds unlocks considerable advancements across diverse scientific fields. Accurate computational descriptions of these compounds are fundamental to designing more effective catalysts, as they allow researchers to predict and optimize reaction mechanisms with greater confidence. In materials science, this improved modeling capability facilitates the discovery and engineering of novel materials possessing tailored electronic, magnetic, and optical properties. Furthermore, a deeper understanding of transition metal compounds is crucial in biochemistry, where these elements play vital roles in metalloenzymes and biological processes; precise modeling can illuminate these mechanisms and aid in the development of new therapeutic strategies. Consequently, this methodological leap promises to accelerate innovation and discovery in these interconnected domains, paving the way for breakthroughs in sustainable energy, advanced materials, and human health.
The singlet-triplet gap, representing the energy difference between the lowest excited singlet state and the lowest excited triplet state, dictates the efficiency of photochemical processes in transition metal compounds. A precise understanding of this gap allows researchers to engineer materials optimized for specific applications; smaller gaps favor intersystem crossing – the process by which a molecule transitions from a singlet to a triplet state – and enhance the efficiency of phosphorescence and photocatalysis. Consequently, manipulating this gap through ligand design or metal selection offers a powerful route to develop highly efficient photocatalysts for solar energy conversion and water splitting, as well as materials exhibiting tailored optoelectronic properties for applications ranging from organic light-emitting diodes to quantum computing. Accurate computational prediction of this crucial property, therefore, accelerates the discovery of novel materials with enhanced performance and functionality.
The accurate prediction of behavior within complex metal centers holds immense promise for advancements in both pharmaceutical development and biological understanding. Many enzymes, crucial for life’s processes, rely on these metal centers to catalyze reactions, and a detailed computational grasp of their electronic structure allows researchers to model enzyme function with increasing fidelity. This capability extends to drug design, where metal-containing compounds are increasingly explored for therapeutic applications – from anti-cancer agents to contrast dyes for medical imaging. By accurately simulating the interactions of these compounds with biological targets, researchers can optimize drug efficacy and minimize potential side effects. Furthermore, understanding how metals participate in essential biological processes – such as oxygen transport in hemoglobin or electron transfer in photosynthesis – requires precise modeling of the metal center’s electronic and structural characteristics, enabling a deeper comprehension of life’s fundamental mechanisms.
Recent computational advancements have yielded a significant leap in the accuracy of predicting metal-metal bond dissociation energies. Utilizing a GGA-referenced CCSD(T) method, researchers have achieved a mean absolute error of only 0.11 to 0.14 eV – a dramatic improvement over traditional HF-CCSD(T) calculations, which exhibit a substantially larger mean absolute error of 0.95 eV. This enhanced precision directly impacts fields reliant on accurate modeling of these bonds, offering the potential for more reliable predictions in areas such as catalyst design, materials science, and the development of novel chemical processes where understanding bond strengths is paramount. The reduced error margin signifies a substantial step towards in silico material discovery and optimization, paving the way for targeted design with minimized experimental trial and error.
The continued development of the KS-CC method represents a promising avenue for tackling increasingly challenging systems in computational chemistry. Researchers are now directing efforts toward extending its applicability to larger, more intricate molecular structures, pushing the boundaries of what is computationally feasible. Simultaneously, investigations are underway to incorporate more sophisticated correlation treatments – beyond the current level of theory – to further refine the accuracy of predicted properties. This includes exploring techniques that account for dynamical correlation effects with greater precision, ultimately aiming to minimize discrepancies between theoretical predictions and experimental observations. Such advancements will not only enhance the reliability of computational models but also unlock new possibilities for materials discovery, catalyst design, and the fundamental understanding of chemical processes.
The pursuit of accurate electronic structure calculations, as demonstrated in this research, inherently involves challenging established boundaries. The study’s successful integration of Kohn-Sham density functional theory with coupled cluster methods exemplifies a willingness to dissect and rebuild existing frameworks. It isn’t merely about achieving greater precision, but about redefining the starting point for complex calculations. This resonates with the sentiment expressed by Werner Heisenberg: “The very act of observing changes that which you seek to observe.” Just as Heisenberg’s uncertainty principle highlights the observer’s impact on a quantum system, this research reveals how a refined initial density can fundamentally alter the outcome of a coupled cluster calculation, particularly for systems exhibiting multireference character. The work underscores that knowledge isn’t passively received, but actively constructed through iterative refinement and, at times, deliberate disruption of conventional approaches.
Beyond the Density: Future Directions
The successful marriage of density functional theory and coupled cluster methods presented here doesn’t resolve the inherent ambiguities within electronic structure calculations-it merely shifts the locus of approximation. The research illuminates a pragmatic solution for systems where traditional wavefunction methods falter, but begs the question: how robust is this ‘rescue’ across the periodic table? The reliance on a Kohn-Sham density as a starting point introduces a dependence on the functional itself – a dependency that demands systematic investigation. One anticipates a future defined by the refinement of density functionals specifically tailored to serve as optimal CC anchors, effectively trading one set of errors for another, but with demonstrably improved accuracy for challenging cases.
The true test lies in extending this approach beyond thermochemistry. Excited state properties, magnetic resonance parameters, and even catalytic reaction mechanisms remain largely unexplored territory. The method’s scalability will also prove critical; while the presented calculations demonstrate feasibility, the computational cost associated with large, complex molecules could quickly become prohibitive. A deeper understanding of the density difference analysis, and how it correlates with the underlying multireference character, might reveal predictive power – allowing for a priori selection of systems where this density-encoded CC will provide the greatest benefit.
Ultimately, this work serves as a reminder that electronic structure theory is not about ‘solving’ the Schrödinger equation, but about building increasingly sophisticated approximations. The black box remains largely unopened, but each successful manipulation-each clever trick-offers a glimpse into the complex machinery within.
Original article: https://arxiv.org/pdf/2602.06149.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- All Skyblazer Armor Locations in Crimson Desert
- One Piece Chapter 1180 Release Date And Where To Read
- How to Get the Sunset Reed Armor Set and Hollow Visage Sword in Crimson Desert
- All Shadow Armor Locations in Crimson Desert
- All Golden Greed Armor Locations in Crimson Desert
- How to Beat Stonewalker Antiquum at the Gate of Truth in Crimson Desert
- Cassius Morten Armor Set Locations in Crimson Desert
- Grime 2 Map Unlock Guide: Find Seals & Fast Travel
- Amber Alert Secrets & CDs In Crime Scene Cleaner Act 2
- USD RUB PREDICTION
2026-02-09 13:52