Author: Denis Avetisyan
Researchers have developed a powerful computational approach to model how electrons move between molecules, accounting for the complex interplay of spin and relativistic effects.
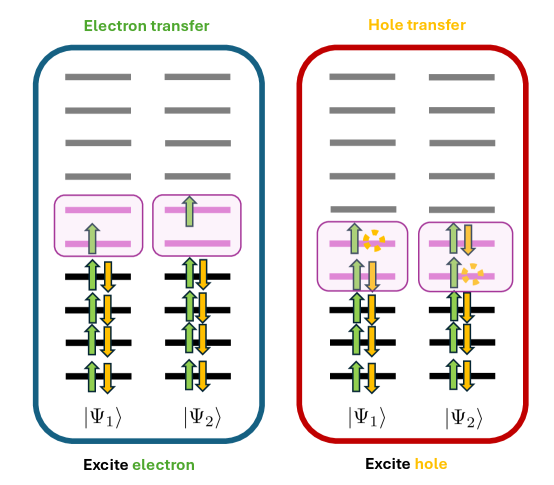
This work presents a generalized complex-valued CASSCF method with a constrained algorithm for investigating charge transfer processes in systems with significant spin-orbit coupling.
Modeling charge transfer-a fundamental process in chemistry and materials science-becomes increasingly challenging when spin-orbit coupling (SOC) and odd electron counts are involved, often leading to complex potential energy surfaces. This work, titled ‘Charge Transfer with a Spin. I: A Generalized CASSCF Framework for Investigating Charge Transfer in the Presence of Spin-Orbit Coupling’, introduces a generalized constrained Complete Active Space Self-Consistent Field (CASSCF) method incorporating complex-valued spinor orbitals to accurately describe these systems. The resulting framework enables smooth potential energy surfaces and rapid convergence even with strong SOC, offering an efficient means to study nonadiabatic dynamics in systems with nontrivial spin degrees of freedom. Will this approach unlock a more detailed understanding of charge transfer in complex chemical and biological environments impacted by relativistic effects?
The Intricacies of Charge Transfer: A Foundation for Understanding Reactivity
Charge transfer, the movement of an electron from one chemical species to another, underpins a vast array of phenomena crucial to life and technology. From photosynthesis and respiration to the operation of solar cells and the design of novel catalysts, this seemingly simple process governs reactivity and energy flow. However, accurately predicting and modeling charge transfer remains a formidable challenge for scientists. The inherent quantum mechanical nature of electron behavior, combined with the complex interplay of molecular structures and environmental factors, necessitates sophisticated computational approaches. Existing methodologies often struggle to capture the subtle nuances of these interactions, particularly in large, biologically relevant systems where multiple pathways and competing processes exist. Consequently, a deeper understanding of the fundamental principles governing charge transfer is paramount to advancing fields ranging from materials science to biochemistry and enabling the design of more efficient and sustainable technologies.
Simulating the dynamics of charge transfer in realistic systems presents a considerable hurdle for conventional computational approaches, particularly when coupled with proton transfer. These methods frequently treat electron and proton movements as separate events, or rely on approximations that break down when they occur in concert – a common scenario within the intricate machinery of biological reactions. Enzymes, for instance, often facilitate reactions by precisely coordinating the transfer of both electrons and protons, demanding a holistic treatment that captures their interconnectedness. The difficulty arises from the vastly different timescales involved; proton transfer is typically much faster than electron transfer, requiring computational techniques capable of accurately resolving both processes simultaneously to provide a complete and reliable depiction of the reaction mechanism.
The efficient capture of solar energy and the acceleration of chemical reactions, both cornerstones of sustainable technology, hinge on the delicate balance of charge and proton transfer processes. In solar cells, the ability to rapidly separate photo-generated electron-hole pairs – a form of charge transfer – and transport them to electrodes dictates overall efficiency. Similarly, catalysts rely on precisely orchestrated proton and electron movements to lower activation energies and speed up desired reactions. Optimizing these coupled transfers requires a deep understanding of how they influence each other; for instance, proton transfer can stabilize charge-separated states, preventing recombination and enhancing performance. Consequently, researchers are increasingly focused on designing materials and systems that specifically manipulate these intertwined processes, aiming to create more effective solar energy conversion and catalytic technologies with broad applications in renewable energy and chemical synthesis.
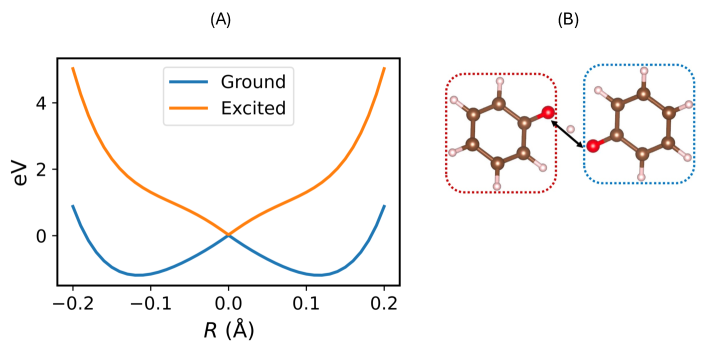
The phenoxy-phenol system offers researchers a uniquely tractable platform for dissecting the intricacies of coupled electron and proton transfer, a process central to numerous chemical and biological functions. This molecular duo exhibits a delicate balance between accepting and donating both electrons and protons, mirroring the behavior observed in more complex scenarios like photosynthesis and enzymatic catalysis. However, accurately modeling this seemingly simple system demands substantial computational power and advanced theoretical approaches. Traditional methods often fall short due to the inherent quantum mechanical effects and the need to simultaneously account for the movements of multiple protons and electrons, necessitating the use of high-level quantum chemical calculations and potentially, molecular dynamics simulations to fully capture the coupled dynamics and unravel the underlying mechanisms driving charge transfer.
Advancing Electronic Structure Methods: A Necessary Precision
The Complete Active Space Self-Consistent Field (CASSCF) method is a quantum chemical technique used to calculate the electronic structure of molecules, particularly those exhibiting strong multi-configurational character. Unlike Hartree-Fock or Density Functional Theory, CASSCF does not rely on a single determinant to approximate the wavefunction; instead, it expands the wavefunction as a linear combination of Slater determinants formed from orbitals within a selected “active space.” This active space comprises the valence orbitals and any core-excited or Rydberg orbitals crucial for describing the electronic behavior of the system. By optimizing both the orbital coefficients and the configuration interaction coefficients simultaneously, CASSCF provides a more accurate representation of electronic correlation effects, enabling the study of bond breaking, excited states, and other complex phenomena where single-determinant approaches fail. The computational cost scales factorially with the size of the active space, limiting its application to systems where the active space can be reasonably sized, typically ranging from 2 to 16 orbitals.
Complete Active Space Self-Consistent Field (CASSCF) calculations scale factorially with the number of active orbitals, rendering them computationally expensive for molecules beyond a certain size. This computational demand arises from the iterative process required to solve the Schrödinger equation within the active space. The Direct Inversion of Iterative Subspace (DIIS) algorithm significantly accelerates convergence by constructing a linear predictor for the wavefunction based on previous iterations. DIIS effectively extrapolates towards the converged solution, reducing the number of iterations required and thus decreasing the overall computational cost. Without DIIS, CASSCF calculations on even moderately sized systems may fail to converge or require impractically long computation times.
The extended and higher-order Density Subspace Correction (eDSC/hDSC) method builds upon the Complete Active Space Self-Consistent Field (CASSCF) approach to improve the accuracy of electronic structure calculations, particularly for systems with unpaired electrons – known as open-shell systems. eDSC/hDSC incorporates symmetry constraints into the CASSCF wavefunction, which reduces the computational space and stabilizes the calculation. This symmetry adaptation, combined with the correction of dynamic correlation effects, results in a more reliable description of the electronic structure and, crucially, generates a smoother potential energy surface. This smoothness is essential for accurate geometry optimizations and simulations of molecular dynamics, as it minimizes numerical instabilities and allows for more reliable predictions of chemical behavior.
Addressing Time-Reversal Symmetry: A Prerequisite for Accurate Open-Shell Systems
Open-shell systems, differing from closed-shell systems which possess all electrons paired, exhibit unique electronic structures arising from the presence of unpaired electrons. This unpaired electron character results in a net electron spin and associated magnetic moment, fundamentally altering the system’s behavior. Consequently, standard computational approaches designed for closed-shell systems – which rely on the assumption of paired spin – become inadequate. The presence of unpaired electrons leads to spatial and spin distributions that necessitate specialized theoretical treatments to accurately describe the system’s electronic structure, energetics, and properties. These systems are prevalent in many areas of chemistry and materials science, including radical chemistry, transition metal complexes, and defects in solids, all of which require tailored computational methods.
Time-reversal symmetry, a cornerstone of quantum mechanics, dictates that the laws of physics remain consistent regardless of time’s direction. In open-shell systems, where unpaired electrons exist, this symmetry is often broken by the inherent asymmetry of the electronic configuration. The Kramers-Restricted Open-Shell (KROS) approach addresses this by explicitly enforcing time-reversal symmetry during calculations. KROS achieves this by constructing wavefunctions that are singlets with respect to a chosen symmetry operator, effectively doubling the size of the Hilbert space and ensuring that all degenerate states are paired. This restriction is crucial for obtaining accurate results, particularly when investigating magnetic properties or performing spectroscopic analyses, as it avoids spurious symmetry breaking and ensures the validity of selection rules.
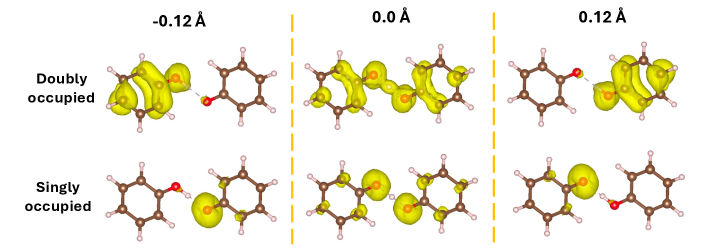
Complex spinors are mathematical entities used in quantum mechanical calculations to represent particles with spin, such as electrons. Unlike simple scalar wavefunctions, complex spinors incorporate both spin-up and spin-down components, and crucially, account for the phase relationship between them. In the context of open-shell systems, where unpaired electrons exist, these spinors are essential for accurately describing the electron’s spin and spatial properties. The use of complex spinors ensures proper treatment of spin-orbit coupling and avoids spurious solutions that can arise when neglecting the full spin information. Specifically, a two-component spinor \begin{pmatrix} \psi_{\uparrow} \\ \psi_{\downarrow} \end{pmatrix} describes the spin state, where \psi_{\uparrow} and \psi_{\downarrow} represent the spin-up and spin-down components, respectively. Their complex nature is fundamental for preserving the correct symmetry properties of the system and obtaining reliable results in electronic structure calculations.
The eDSC/hDSC method implements a symmetry-constrained approach to open-shell calculations by explicitly enforcing time-reversal symmetry, which is critical for accurately representing systems with unpaired electrons. Empirical results demonstrate that the energy gap, representing the difference between singlet and triplet states, scales quadratically with the strength of spin-orbit coupling; specifically, the energy gap \Delta E exhibits a relationship of \Delta E \propto \lambda^2, where λ represents the spin-orbit coupling constant. This quadratic scaling behavior has been consistently observed across a range of open-shell systems and provides a quantifiable metric for assessing the impact of relativistic effects on electronic structure.
Nuclear Momentum and Implications for Charge Transfer: A Complete Depiction of Reactivity
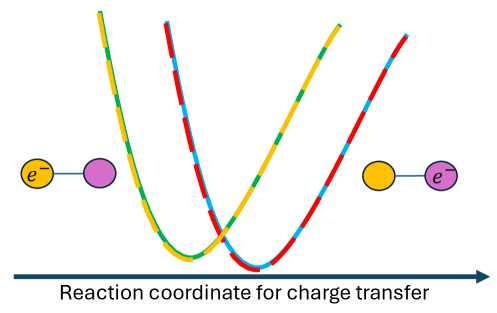
Traditional modeling of charge transfer often prioritizes electronic structure, yet a complete depiction necessitates acknowledging the significant influence of nuclear momentum. The nuclei within a reacting system aren’t static; their motion-their momentum-directly impacts the potential energy surfaces governing the reaction. This momentum alters the energetic landscape, effectively steering the reaction along specific pathways and influencing the probability of successful charge transfer. Ignoring this kinetic component can lead to inaccurate predictions of reaction rates and mechanisms, particularly in systems where light nuclei or significant vibrational excitation are present. Consequently, incorporating a detailed treatment of nuclear momentum provides a more realistic and comprehensive understanding of charge transfer dynamics, revealing nuances often obscured by purely electronic structure-based approaches.
Charge transfer, the movement of electrons between molecular entities, isn’t a static event; its progression through time is fundamentally linked to the motion of the nuclei involved. Traditional models often treat molecules as fixed structures, yet in reality, these systems are constantly vibrating, rotating, and translating – a phenomenon known as nuclear motion. This motion dramatically influences how electrons redistribute during a reaction, creating a dynamic landscape of potential energy surfaces. In dynamic environments – such as those found in solutions or at interfaces – nuclear momentum becomes particularly crucial, as collisions and thermal fluctuations can significantly alter reaction pathways and rates. Accurately capturing this nuclear dynamics is therefore essential for a complete understanding of charge transfer processes, enabling predictions of reaction timescales and efficiencies that static models simply cannot achieve.
A comprehensive understanding of chemical reactions requires a simultaneous consideration of both electronic structure and nuclear momentum. Traditional approaches often prioritize electronic details, yet the motion of atomic nuclei significantly influences how reactants transform into products, particularly during dynamic processes. By accurately modeling electronic interactions and incorporating the full complexity of nuclear motion-including vibrational and rotational energies-researchers can unlock a more complete picture of reaction pathways. This integrated approach allows for precise prediction of reaction rates and the identification of crucial intermediate states, ultimately offering a deeper insight into the fundamental mechanisms governing crucial chemical transformations and providing a foundation for the rational design of new materials and catalysts.
The integration of nuclear momentum into models of charge transfer isn’t merely a refinement of existing theory, but a pathway towards rationally designed materials for sustainable energy technologies. Accurate simulations, incorporating both electronic structure and nuclear dynamics, allow for the prediction of reaction pathways and energetic landscapes with unprecedented detail. This capability is particularly impactful in the development of catalysts, where understanding charge transfer at active sites is crucial for optimizing performance and selectivity. Similarly, in solar cell design, precisely modeling charge separation and transport-processes fundamentally linked to nuclear motion-can unlock strategies for enhancing light-harvesting efficiency and overall device stability. Consequently, a holistic approach promises not only a deeper understanding of fundamental chemical processes but also a tangible toolkit for accelerating the transition towards cleaner, more efficient energy solutions.
The pursuit of accurately modeling charge transfer, as detailed in this framework, demands a rigorous mathematical foundation. The authors’ development of a generalized CASSCF approach, capable of handling spin-orbit coupling, reflects this need for provable, complete solutions. It’s not simply about achieving numerical results, but about ensuring the underlying theory is sound. As Ernest Rutherford observed, “If you can’t explain it simply, you don’t understand it well enough.” This principle directly applies to the complex calculations presented; the elegance of the method lies in its ability to distill the intricacies of electron transfer into a logically consistent and verifiable model, even when dealing with the complexities introduced by spin and relativistic effects.
Beyond the Transfer
The presented framework, while a demonstrable advance in treating charge transfer with concomitant spin considerations, merely illuminates the extent of what remains unknown. The elegance of a formally correct, complex-valued CASSCF treatment does not negate the inherent approximations within the method itself. One must acknowledge that the true potential energy surface, especially in systems dominated by relativistic effects, remains a construct, an idealization of a reality perpetually beyond complete capture. The efficiency gained through constrained algorithms is, ultimately, a pragmatic concession, not a fundamental solution.
Future development cannot solely focus on extending the applicability to larger systems or more complex chemical scenarios. A critical path lies in rigorously quantifying the impact of basis set incompleteness and the truncation of many-electron operators on the accuracy of predicted charge transfer rates. If a result cannot be reproduced to a defined precision, even within the confines of the chosen theoretical model, its practical value diminishes rapidly.
The ultimate challenge remains the seamless integration of this level of theoretical detail with nonadiabatic dynamics simulations capable of accurately describing decoherence and environmental effects. A mathematically pure description of the initial state is insufficient; the evolution of that state must be equally well-defined, and demonstrably free from artifacts introduced by numerical approximations. Only then can one begin to approach a truly predictive understanding of these fundamental processes.
Original article: https://arxiv.org/pdf/2602.07746.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- All Shadow Armor Locations in Crimson Desert
- The Limits of Thought: Can We Compress Reasoning in AI?
- Genshin Impact Dev Teases New Open-World MMO With Realistic Graphics
- Sega Reveals Official Sonic Timeline: From Prehistoric to Modern Era
- Where to Pack and Sell Trade Goods in Crimson Desert
- ARC Raiders Boss Defends Controversial AI Usage
- Gold Rate Forecast
- Who Can You Romance In GreedFall 2: The Dying World?
- Best Weapons, Armor, and Accessories to Get Early in Crimson Desert
- How to Beat Antumbra’s Sword (Sanctum of Absolution) in Crimson Desert
2026-02-10 22:03