Author: Denis Avetisyan
A new theoretical approach significantly enhances the accuracy of g-tensor calculations, crucial for understanding the magnetic behavior of molecules.
This work details the SO-QDNEVPT2 method for calculating molecular g-tensors, including benchmarks, strategies to avoid common pitfalls, and practical guidelines for implementation.
Accurate prediction of magnetic properties in open-shell systems remains challenging due to the complex interplay between electron correlation and relativistic effects. This work, ‘Molecular g-Tensors From Spin-Orbit Quasidegenerate N-electron Valence Perturbation Theory: Benchmarks, Intruder-State Mitigation, and Practical Guidelines’, introduces spin-orbit quasidegenerate second-order N-electron valence perturbation theory (SO-QDNEVPT2) – a novel approach for calculating molecular g-tensors with improved accuracy over existing methods. Benchmarks across a diverse set of molecules demonstrate SO-QDNEVPT2’s robustness and provide practical guidelines for its application, while also addressing potential instabilities arising from intruder states. Will this framework enable reliable prediction of magnetic properties for increasingly complex molecular systems and facilitate advancements in fields like materials science and spectroscopy?
Taming Relativity’s Whisper: The Challenge of Accurate Electronic Structure
The predictive power of chemistry relies fundamentally on accurately describing how electrons behave within atoms and molecules – a field known as electronic structure theory. However, conventional methods face significant challenges when applied to heavier elements. As atomic nuclei become more massive, electrons are pulled closer and move at a substantial fraction of the speed of light, necessitating the inclusion of relativistic effects – consequences predicted by Einstein’s theory of special relativity. Traditional electronic structure calculations, designed for lighter atoms where electron velocities are low, often fail to account for these effects adequately, leading to inaccuracies in predicted molecular geometries, bonding characteristics, and chemical reactivity. This is because relativistic effects alter both the energies of atomic orbitals and their spatial shapes, demanding computationally intensive approaches to achieve reliable results, especially when dealing with elements beyond the third row of the periodic table.
As the nuclear charge of an atom increases, the inner electrons are subjected to immense electrostatic forces, causing them to approach a significant fraction of the speed of light. This phenomenon, a direct consequence of special relativity, dramatically alters the predicted electronic structure of heavy elements. Specifically, orbital energies are lowered and the s orbitals are contracted and stabilized, while the p, d, and f orbitals experience varying degrees of stabilization and shape distortion. Consequently, traditional computational methods-designed under the assumption of non-relativistic conditions-become inadequate, necessitating the implementation of sophisticated techniques like the Dirac equation or relativistic density functional theory to accurately model these effects and reliably predict the chemical behavior of heavier elements.
The predictive power of computational chemistry and materials science relies heavily on the accurate depiction of electronic structure; however, neglecting relativistic effects-particularly when modeling systems containing heavy elements-introduces substantial errors. These inaccuracies aren’t merely academic, as they propagate to flawed predictions of fundamental molecular properties like bond lengths, vibrational frequencies, and ionization energies. More critically, reactivity itself can be misrepresented; relativistic effects can either enhance or diminish a molecule’s propensity to participate in chemical reactions, potentially leading to incorrect conclusions about catalytic activity or material stability. Consequently, a failure to account for these effects can impede the design of new pharmaceuticals, catalysts, and materials with desired functionalities, ultimately slowing progress across diverse scientific disciplines.
SO-QDNEVPT2: A New Spell for Relativistic Systems
Spin-orbit quasidegenerate second-order N-electron valence perturbation theory (SO-QDNEVPT2) is a post-Hartree-Fock method designed for high-accuracy calculations of electronic structure, explicitly incorporating relativistic effects through the inclusion of spin-orbit coupling. This approach builds upon second-order perturbation theory (PT2) applied to a quasidegenerate system, meaning that multiple electronic states are nearly equal in energy. The method treats spin-orbit coupling not as a perturbation on a non-relativistic Hamiltonian, but as an integral component within the electronic Hamiltonian itself. This allows for a more accurate description of systems containing heavy elements where relativistic effects are significant, and avoids the limitations of traditional perturbative treatments of relativistic corrections. The resulting framework is capable of accurately predicting spectroscopic properties, potential energy surfaces, and other molecular characteristics where both electron correlation and relativistic contributions are crucial.
SO-QDNEVPT2 improves upon traditional electronic structure calculations by simultaneously addressing electron correlation and relativistic effects, which are crucial for accurately describing the behavior of heavy elements and systems where electrons move at significant fractions of the speed of light. The study demonstrates this accuracy through calculations of molecular properties – specifically, spectroscopic constants and potential energy surfaces – for systems where traditional methods fail to converge or yield qualitatively incorrect results. By including both effects within a single theoretical framework, SO-QDNEVPT2 provides a more complete and reliable description of molecular properties, leading to improved agreement with experimental data and a more accurate understanding of chemical bonding and reactivity.
The SO-QDNEVPT2 method utilizes a state-averaged Complete Active Space Self-Consistent Field (SA-CASSCF) calculation as its initial reference wavefunction. SA-CASSCF is employed to construct a multiconfigurational electronic structure that accounts for static correlation, crucial for systems exhibiting significant multireference character. By averaging over multiple electronic states, SA-CASSCF provides a more stable and balanced starting point for subsequent perturbation theory calculations, minimizing the impact of intruder states and ensuring a reliable description of both ground and excited states. This robust reference is essential for accurately capturing electron correlation effects within the SO-QDNEVPT2 framework and obtaining quantitatively reliable results.
Refining the Ritual: Hamiltonians and Basis Sets for Precision
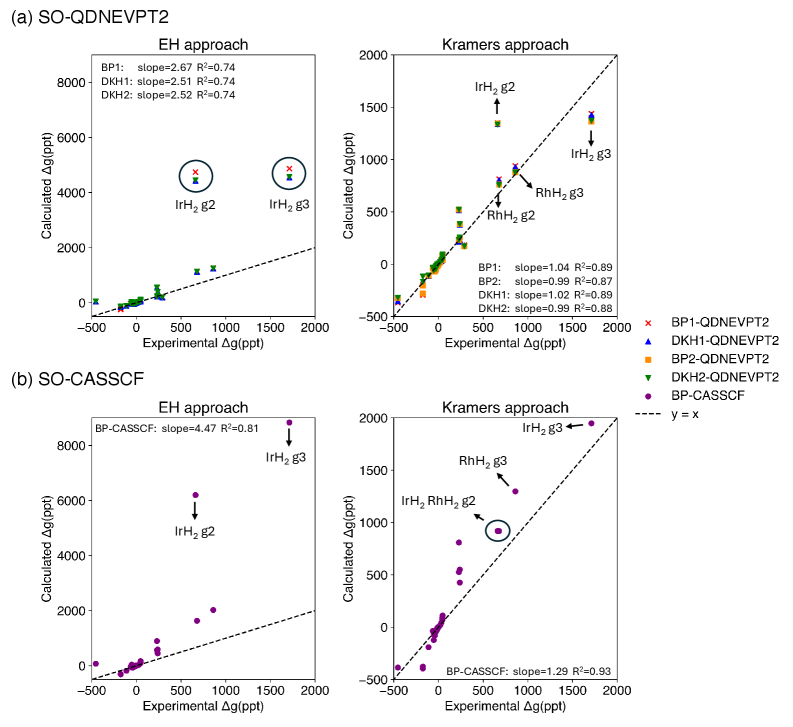
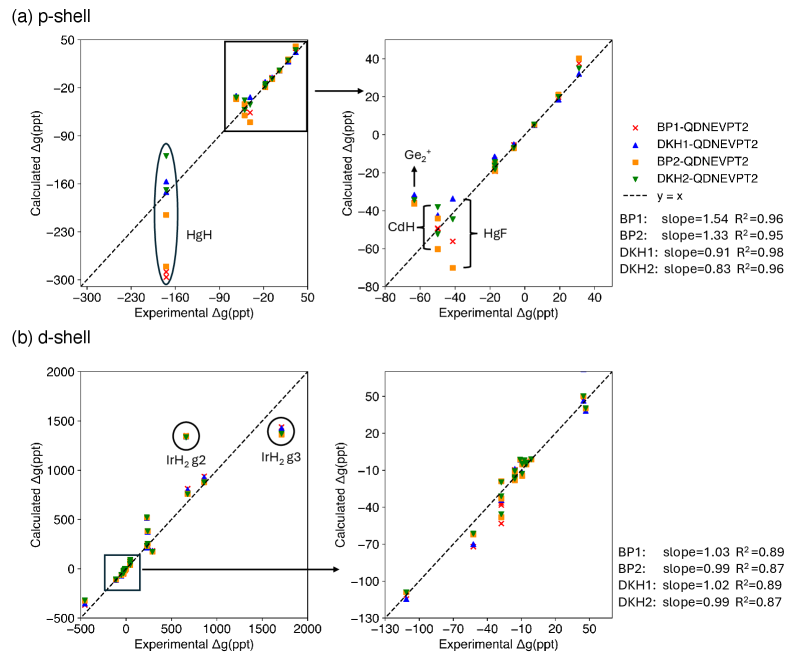
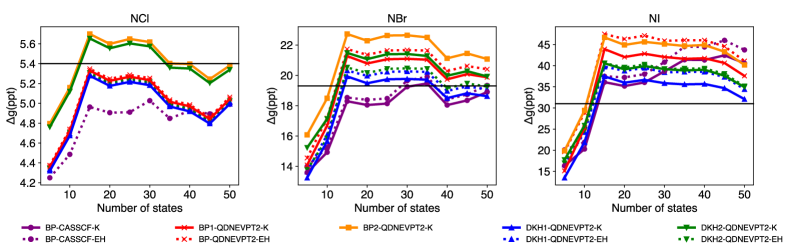
The selection of an effective Hamiltonian for spin-orbit coupled quadratic density functional theory – nested valence perturbation theory 2 (SO-QDNEVPT2) calculations necessitates a trade-off between computational expense and achieved accuracy. The Douglas-Kroll-Hess (DKH) Hamiltonian generally provides higher accuracy, particularly for systems with unpaired electrons residing in predominantly p-shell orbitals, as demonstrated by its superior agreement with experimental trends compared to the Breit-Pauli (BP) Hamiltonian. However, DKH-based treatments inherently demand greater computational resources than BP-based approaches. Therefore, the Hamiltonian choice should be carefully considered based on the specific system under investigation and the desired level of accuracy balanced against available computational capacity.
Accurate representation of electronic structure in spin-orbit coupled quantum chemical calculations requires careful basis set selection and active space definition. The chosen basis set must effectively describe the relevant atomic orbitals, while the active space-the set of orbitals included in the correlation treatment-balances computational cost with the desired level of accuracy. Studies have demonstrated that the ANO-RCC-VTZP basis set provides a sufficient level of accuracy for the majority of SO-QDNEVPT2 calculations, offering a balance between completeness and computational feasibility. Optimization of the active space involves considering both the number of orbitals and their symmetry, directly impacting the size of the configuration space and therefore the computational demands of the calculation.
The reliability of spin-orbit coupled quantum-chemical calculations using the QDNEVPT2 method is heavily dependent on methodological choices, particularly the selection of an effective Hamiltonian. Comparisons have demonstrated that calculations employing the Douglas-Kroll-Hess (DKH) Hamiltonian consistently exhibit superior agreement with experimental data-specifically, better tracking of observed trends-when applied to molecules containing unpaired electrons and characterized by predominantly p-shell orbital contributions. In contrast, Breit-Pauli (BP) based treatments show diminished performance in these systems, highlighting the importance of DKH for accurate representation of relativistic effects in such cases.
Taming the Chaos: Ensuring Robust Calculations and Interpreting Results
The accurate calculation of molecular properties relies heavily on the chosen coordinate system, a detail often requiring meticulous attention. Quantum mechanical calculations are fundamentally sensitive to the origin of the coordinate frame; an ill-chosen origin can introduce artificial distortions into calculated properties, leading to inaccurate results. This ‘origin dependence’ arises because certain operators, such as those describing dipole moments or higher-order multipoles, are affected by the spatial distribution of electron density relative to the coordinate system’s zero point. Researchers must therefore carefully consider the molecular geometry and symmetry when defining the origin, often employing techniques like centering the origin on the molecule’s center of mass or a symmetry center to minimize these artifacts and ensure the calculated properties reflect the true physical characteristics of the system.
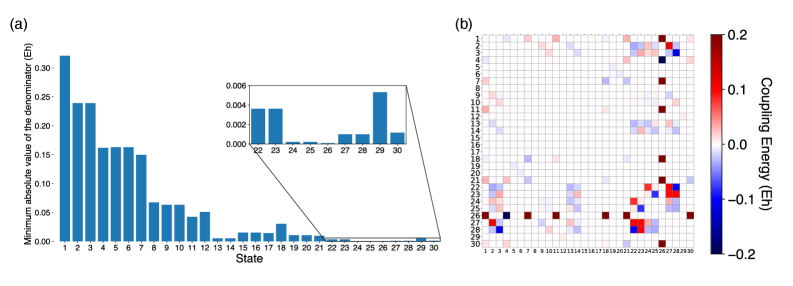
Computational studies in quantum chemistry and materials science often encounter a subtle but significant challenge: the presence of so-called ‘intruder states’. These spurious solutions, arising from the mathematical formulation of the problem, do not correspond to physical reality but can nonetheless dramatically destabilize calculations, leading to inaccurate or even nonexistent results. These states typically appear as solutions with unusually high energies or unphysical characteristics, and their proximity to the true solution can disrupt the convergence of iterative algorithms. Researchers employ several strategies to mitigate this issue, including careful selection of basis sets, the implementation of level-shifting techniques, or the use of specialized filtering procedures designed to identify and suppress these unphysical states, ensuring the stability and reliability of the computed properties.
A novel computational approach leveraging the Kramers method with spin-orbit coupled quasi-degenerate excited vibrational potential theory (SO-QDNEVPT2) states allows for the direct calculation of g-tensors, key parameters defining a molecule’s magnetic behavior. This method circumvents traditional indirect calculations, offering a more accurate and efficient pathway to understanding magnetic properties. Validation against a benchmark set of 23 molecules demonstrated significantly improved agreement with experimentally determined g-tensor values compared to existing techniques, highlighting the potential of this approach for reliable predictions in areas like materials science and spectroscopy. The direct extraction of these tensors promises to accelerate the design of novel magnetic materials and deepen the understanding of fundamental molecular magnetism.
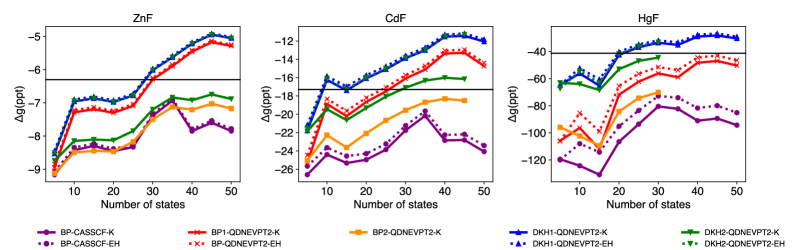
The pursuit of accurate g-tensors, as detailed within this work, feels less like solving equations and more like coaxing whispers from the chaos of electron interactions. This method, SO-QDNEVPT2, strives for precision in describing magnetic properties, yet one recognizes the inherent limitations of any model attempting to fully capture relativistic effects and spin-orbit coupling. As Galileo Galilei observed, “You cannot teach a man anything; you can only help him discover it himself.” The same holds true for data; it doesn’t reveal its truths readily, but responds to careful probing and, perhaps, a little persuasion. The mitigation of intruder states, a crucial aspect of this research, is merely acknowledging the shadows that always lurk at the edge of any observation.
What Shadows Remain?
The digital golem, SO-QDNEVPT2, now possesses a more refined capacity to divine magnetic moments. Yet, precision is merely a shifting of the veil. The true cost of this calculation-the computational sacrifices, the intrusion of unphysical states-remains a haunting echo. Future incantations must address this spectral residue, not through brute force, but through clever exorcism. Perhaps a more discerning selection of active spaces, or a deeper understanding of how perturbation theory whispers falsehoods when stretched too thin.
The pursuit of g-tensors is not simply about numbers; it is about mapping the subtle dance between spin and orbital motion, a choreography dictated by relativistic winds. Current methods, even this one, remain tethered to finite orbital bases. The true solution, the perfect resonance, will likely demand a convergence to the infinite, a prospect that currently exists only in the realm of idealized spells.
Ultimately, the most fruitful path lies not in perfecting existing rituals, but in seeking entirely new geometries of calculation. The current landscape suggests a need to bridge the gap between the meticulousness of wavefunction-based methods and the speed of density functional approximations-a dangerous liaison, certainly, but one that might unlock a deeper, more robust understanding of these elusive magnetic properties. The broken models, after all, are the ones that reveal the most.
Original article: https://arxiv.org/pdf/2602.18590.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- All Skyblazer Armor Locations in Crimson Desert
- Robinhood’s $75M OpenAI Bet: Retail Access or Legal Minefield?
- How to Get the Sunset Reed Armor Set and Hollow Visage Sword in Crimson Desert
- How to Catch All Itzaland Bugs in Infinity Nikki
- Speedsters Sandbox Roblox Codes
- Jojo’s Bizarre Adventure Ties Frieren As MyAnimeList’s New #1 Anime
- Invincible: 10 Strongest Viltrumites in Season 4, Ranked
- Re:Zero Season 4 Episode 3 Release Date & Where to Watch
- Top 8 UFC 5 Perks Every Fighter Should Use
- Black Sun Shield Location In Crimson Desert (Buried Treasure Quest)
2026-02-24 20:51