Author: Denis Avetisyan
Researchers have developed a refined method for representing and compressing sets of genomic strings, offering improved efficiency and accuracy in bioinformatics applications.
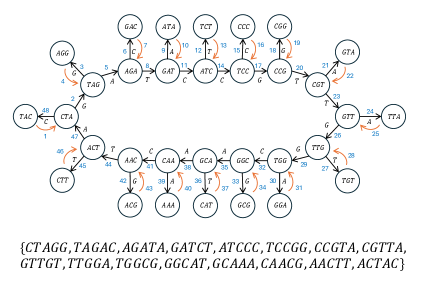
This work introduces ‘necklace covers’ of de Bruijn graphs as a novel technique for identifying spectrum-preserving string sets, achieving comparable or superior compression to existing methods while ensuring exactness and avoiding false positives.
Efficiently storing and accessing k-mer spectra is critical in computational genomics, yet existing methods face limitations in representing complex relationships within genomic data. This paper, ‘Variations on the Problem of Identifying Spectrum-Preserving String Sets’, introduces ‘necklace covers’-a novel framework for representing k-mer sets as branching structures on de Bruijn graphs. Experimental results demonstrate that this approach achieves comparable or superior compression to existing techniques like Eulertigs and Masked Superstrings while guaranteeing exactness in k-mer spectrum preservation. Could this new representation paradigm unlock further advancements in genome assembly and analysis by more effectively capturing the intricacies of genomic variation?
The Inevitable Bottleneck: Representing the Genome
Genomic data analysis fundamentally relies on representing the vast sequences of nucleotides, often broken down into shorter subsequences called k-mers. This approach, while powerful, quickly becomes computationally expensive due to the sheer scale of genomic datasets. Consider that a relatively modest genome contains billions of base pairs; enumerating and storing all possible k-mers, even with a small k value, demands substantial memory and processing power. The combinatorial explosion of potential k-mers necessitates innovative data structures and algorithms to manage this complexity, as traditional methods often struggle to efficiently represent and access these sequences without incurring significant redundancy or slowing down analytical pipelines. Effectively handling k-mer representation is therefore a critical bottleneck in modern genomics, directly impacting the feasibility of large-scale studies and the speed of crucial analyses.
Existing approaches to storing genomic data often face a fundamental trade-off: achieving a highly compact representation frequently hinders efficient data access, while prioritizing speed and ease of retrieval typically introduces significant redundancy. Methods like simple string storage consume excessive memory, particularly with the massive scale of modern genomic datasets, but compressed formats can demand substantial computational resources for decompression and random access. The challenge lies in developing data structures that minimize storage space-critical for handling increasingly large genomes-without sacrificing the ability to quickly locate and retrieve specific k-mers or sequence fragments. This balancing act is paramount because slow access times can severely bottleneck downstream analyses, impacting the feasibility of tasks like genome assembly, variant calling, and comparative genomics.
A central challenge in genomics lies in devising data structures capable of faithfully representing the immense volume of sequence information while simultaneously minimizing computational demands. Researchers strive for a balance between accurate replication of the original genetic code and practical considerations of memory usage and processing speed. These structures must not only store the k-mer sequences but also allow for rapid retrieval and manipulation, enabling efficient downstream analyses such as genome assembly, where fragmented sequences are pieced together, and variant calling, which identifies differences within the genome. The development of such optimized data representations is thus pivotal for advancing genomic research, as it directly impacts the feasibility and scalability of complex computational tasks.
The utility of genomic data fundamentally rests on its accurate and efficient representation, directly impacting the success of complex downstream analyses. Genome assembly, the process of reconstructing a complete genome from fragmented reads, demands a data structure that minimizes memory footprint while enabling rapid comparisons and overlaps between k-mers. Similarly, variant calling – identifying genetic differences contributing to disease or adaptation – relies on the ability to precisely map reads to a reference genome and detect subtle variations; any inaccuracies or inefficiencies in the underlying data representation can lead to false positives or missed true variants. Consequently, optimized genomic representations aren’t merely about storage; they are the bedrock upon which meaningful biological insights are built, shaping the reliability and interpretability of genomic research.
From Fragmentation to Coherence: Building Efficient Structures
Spectrum-Preserving String Sets (SPSS) are a data compression technique used in bioinformatics to represent a set of strings, typically k-mers from genomic data, in a compact form. The core principle of SPSS involves storing only the minimal information needed to reconstruct the original set of strings, thereby reducing memory usage. While SPSS effectively preserves the original spectrum of strings, its initial implementations can contain redundant information. This redundancy arises from the storage of strings that are subsets of others, or from storing equivalent representations. Subsequent optimization techniques, such as Simplitigs, directly address these inefficiencies by systematically eliminating redundant entries and refining the data structure to achieve a more memory-efficient representation without compromising the ability to reconstruct the original string set.
Simplitigs represent an optimization of Spectrum-Preserving String Sets (SPSS) achieved through the removal of redundant k-mers. While SPSS aim to retain all possible k-mers within a sequence, this can lead to significant memory consumption, especially with repetitive genomes. Simplitigs address this by identifying and storing only the unique k-mers, effectively collapsing identical sequences into a single representation. This reduction in data storage is accomplished by replacing redundant k-mers with a count of their occurrences, resulting in a more compact data structure without losing the ability to reconstruct the original sequence. The efficiency gain is directly proportional to the degree of redundancy present in the input data; highly repetitive sequences benefit most from simplitig representation.
Necklace Covers represent data as cycles and paths within a graph structure, offering advantages for efficient data traversal. This approach models the data as a ‘necklace’ where each element is a node, and connections between nodes represent relationships or sequences. Cycles within the graph denote repeating patterns, while paths represent unique sequences. By representing data in this manner, algorithms can efficiently navigate the dataset by following these defined paths and cycles, reducing the need for redundant comparisons or exhaustive searches. This is particularly useful in applications involving sequence analysis or pattern matching, where identifying and traversing specific patterns is crucial for performance.
De Bruijn graphs provide a method for representing and efficiently storing sequences of data, particularly in genomics where short, overlapping subsequences, termed kk-mers, are common. A de Bruijn graph is constructed by representing each unique kk-mer as a node and drawing a directed edge between nodes if the (k-1)-suffix of the first kk-mer is equal to the (k-1)-prefix of the second. This graph structure inherently captures overlaps between kk-mers, eliminating redundant storage of shared sequences. Traversal of this graph, either as paths or cycles, effectively reconstructs the original sequence while requiring space proportional to the number of unique kk-mers rather than the total length of the sequence, leading to significant memory savings in compact representation schemes like Simplitigs and Necklace Covers.
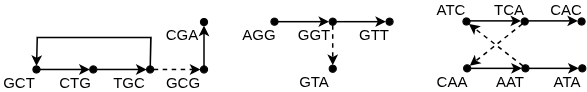
The Geometry of Minimal Coverage: Algorithms in Action
Eulertigs represent a technique for constructing a minimal Single-Path String Packing (SPSS) by leveraging Eulerian tours within a de Bruijn graph. A de Bruijn graph, constructed from the input strings, represents all possible overlapping k-mers. An Eulerian tour, a path that visits every edge exactly once, can be extracted if the graph is Eulerian or can be made Eulerian by adding minimal edges. This tour directly corresponds to a string visiting all input strings. The resulting string, or Eulertig, provides a compact representation of the original strings, minimizing the number of paths needed to cover all input sequences and therefore forming a minimal SPSS. The efficiency of this method relies on the ability to efficiently find Eulerian tours within the de Bruijn graph.
A minimum Path-Cycle (PC) cover, in the context of minimal Shortest Path Spanning (SPSS) algorithms, represents the smallest set of node-disjoint paths and cycles required to include every node within the de Bruijn graph. The objective of finding this cover is to minimize the total length of these paths and cycles, thereby optimizing the resulting SPSS. Each node must belong to exactly one path or cycle within the cover; therefore, the cover constitutes a complete partitioning of the graph’s node set. Algorithms designed to find minimum PC covers are crucial for efficiently constructing minimal SPSS solutions, as the size and length of the cover directly impact the overall performance and resource usage of the spanning set.
Determining a minimum Path-Cycle (PC) cover within a de Bruijn graph can be efficiently achieved through the application of bipartite matching algorithms. The nodes of the graph are partitioned into two sets, representing the starting and ending nodes of potential paths and cycles. An edge connects a starting node to an ending node if a corresponding edge exists in the de Bruijn graph. A maximum bipartite matching on this derived graph directly corresponds to a maximum set of edges covered by paths and cycles. The remaining uncovered edges then define the minimum number of cycles required to complete the PC cover, ensuring all nodes are visited with minimal redundancy. This approach leverages well-established polynomial-time algorithms, such as the Hopcroft-Karp algorithm, for optimal performance in finding the minimum PC cover.
Greedy Depth-First Search (DFS) provides a heuristic method for constructing necklace covers in de Bruijn graphs, offering a computationally efficient alternative to exact minimum PC cover algorithms. Unlike algorithms guaranteeing optimality, the greedy DFS approach iteratively extends paths by selecting the next available node based on immediate connectivity, prioritizing traversal without backtracking unless absolutely necessary. While this method doesn’t guarantee the absolute smallest cover, its linear time complexity – typically O(n), where n represents the number of nodes in the graph – makes it suitable for large-scale sequence assembly problems where finding a near-optimal solution within a reasonable timeframe is prioritized over absolute minimization. The resulting necklace cover, though potentially redundant, provides a complete path covering of the input graph and is often sufficient for practical applications.
The Shadow of Approximation: Accuracy and its Cost
Compact representations of genomic data, such as those employing Masked Superstrings, prioritize efficiency by minimizing storage space; however, this compression can inadvertently introduce false positive k-mers – sequences that appear within the representation but were not originally present in the biological sample. This occurs because the algorithm reconstructs strings, and in doing so, may generate novel k-mers not found in the source data. While advantageous for reducing memory footprint, the presence of these spurious k-mers poses a challenge for downstream analyses like sequence alignment or species identification, potentially leading to inaccurate results and requiring careful consideration of mitigation strategies or the implementation of filtering steps to remove these artifacts.
The introduction of spurious k-mers, termed false positives, poses a significant challenge to the reliability of analyses relying on compact sequence representations. These artifacts, while stemming from the efficiency of methods like Masked Superstring, can lead to inaccurate conclusions in downstream processes such as genome assembly, taxonomic classification, and variant calling. For instance, a false positive might be incorrectly identified as a unique genetic marker or contribute to misinterpretations of species abundance. Consequently, researchers must carefully evaluate the potential for false positives and implement mitigation strategies, including filtering based on sequence abundance, utilizing complementary data sources for validation, or employing algorithms designed to minimize their occurrence, ensuring the integrity of subsequent biological inferences.
The methodology introduces Parenthesis Representation as a powerful visualization tool for necklace covers, offering a structural depiction that greatly simplifies debugging and analytical processes. This representation employs balanced parentheses to mirror the cyclical relationships within the k-mer spectrum, allowing researchers to intuitively trace the connections between different sequence fragments. By visually encoding the cover’s structure, potential errors or inconsistencies become readily apparent, enabling efficient identification and correction of issues that might otherwise remain hidden within complex datasets. This approach facilitates a deeper understanding of the underlying sequence composition and improves the reliability of downstream analyses by providing a clear, interpretable framework for data validation and quality control.
A newly developed method for representing genomic sequences, based on a ‘necklace’ structure, demonstrably rivals and often surpasses the space efficiency of techniques like Masked Superstring, particularly as the k-mer size increases. Crucially, this representation doesn’t merely compress data-it preserves the complete k-mer spectrum of the original sequence without introducing spurious false positives. This exact preservation is a significant advantage, as it ensures downstream analyses aren’t skewed by artificially generated data. By organizing k-mers into interconnected ‘necklaces’, the approach achieves a highly compact representation, effectively mirroring the original sequence’s complexity while minimizing storage demands and computational overhead.
For certain input string sets, the novel necklace-based representation exhibits a significant compression advantage over traditional Eulertigs. Specifically, this methodology achieves a reduction in representation size quantified as 4/(k+1), where ‘k’ denotes the length of the k-mers being analyzed. This improved space occupancy is particularly beneficial when dealing with large genomic datasets or complex biological sequences, offering substantial savings in storage and computational resources. The mathematical relationship highlights that as the k-mer length increases, the efficiency gain relative to Eulertigs also grows, demonstrating the scalability and potential of this approach for increasingly complex genomic investigations.
The pursuit of efficient data representation, as demonstrated by this work on spectrum-preserving string sets, reveals a fundamental truth about complex systems. One anticipates eventual limitations, even in the most elegant of constructions. As John McCarthy observed, “A system that never breaks is dead.” This paper’s exploration of necklace covers within de Bruijn graphs isn’t about achieving a perfect, static solution to kk-mer compression. Rather, it’s an acknowledgement that any representation will inevitably face the challenge of evolving data and the need for adaptable structures. The very act of seeking improved compression, while maintaining exactness, implies an acceptance of future refinement – a cycle of building, testing, and inevitably, rebuilding.
What Lies Ahead?
The construction of spectrum-preserving string sets via necklace covers offers a predictable refinement – improved compression ratios will inevitably be pursued. However, the true challenge isn’t squeezing more data into less space; it’s acknowledging the inherent ephemerality of information. Any compression scheme, however ‘exact’, is merely a temporary reprieve from entropy. The system described here, while demonstrably effective, will ultimately succumb to the same pressures as its predecessors – the ever-increasing volume of k-mer data and the inevitable obsolescence of current hardware.
The emphasis on de Bruijn graphs, while currently productive, may prove a local maximum. The graph itself is a simplification, a human imposition of order onto a fundamentally chaotic process. Future work will likely focus not on more efficient graph representations, but on methods that embrace the inherent ambiguity of biological sequences – systems that allow for controlled approximation and probabilistic inference. A guarantee of perfect reconstruction is just a contract with probability, after all.
Stability is merely an illusion that caches well. The exploration of alternative topologies, perhaps inspired by non-Euclidean geometries or network theory, holds a distant promise. The field will not be defined by solving the ‘problem’ of k-mer compression, but by recognizing that the problem is not static – it is the evolution of the data itself. Chaos isn’t failure – it’s nature’s syntax.
Original article: https://arxiv.org/pdf/2602.19408.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- All Skyblazer Armor Locations in Crimson Desert
- Robinhood’s $75M OpenAI Bet: Retail Access or Legal Minefield?
- How to Get the Sunset Reed Armor Set and Hollow Visage Sword in Crimson Desert
- How to Catch All Itzaland Bugs in Infinity Nikki
- Speedsters Sandbox Roblox Codes
- Jojo’s Bizarre Adventure Ties Frieren As MyAnimeList’s New #1 Anime
- Invincible: 10 Strongest Viltrumites in Season 4, Ranked
- Re:Zero Season 4 Episode 3 Release Date & Where to Watch
- Top 8 UFC 5 Perks Every Fighter Should Use
- Black Sun Shield Location In Crimson Desert (Buried Treasure Quest)
2026-02-25 05:25